The frozen secret of proteins
To understand in detail how proteins and other macromolecules function in our tissues and cells, we need to map their spatial structures and interactions. Cryo-electron microscopy has recently become suitable for exploring this grid at the level of atoms and has also proven to be extremely effective on a wide scale. Using a special microscope, the protein molecules are studied at a temperature below -137°C, which makes the inner movements and interactions detectable.
Vitrification (rapid plunge freezing) preserves and fixes the natural structure of macromolecules,
ideally, in many different orientations, as the molecules are rotating in the solution. The electron beam interacts with the studied sample and as a result of processing the images created by its scattering, the 3D shapes (or their projections in 2D) characteristic of the molecule can be arranged into groups. The next step is imaging whereby – similar to 3D scanning used in the macroscopic world – the spatial form of an object can be reconstructed from images taken from several different directions.
The cryo-EM method, recognised by the Nobel Prize in 2017, is suitable for mapping the 3D structures and molecular interactions of large molecular structures, whether viruses or ribosomes, proteasomes, and other protein multimers and complexes, and even for supporting the development of active substances. The ELKH-ELTE Protein Modelling Research Group – Laboratory of Structural Chemistry and Biology, which is among the top 50 research centres on the list of the National Research, Development, and Innovation Office (NKFIH), also uses the method that opens up new possibilities in the field of research into the structure of large proteins.
ELTE scientists previously determined the spatial structure of the AAP (acylaminoacyl peptidase) enzyme tetramer, the form of which was previously unknown in mammals. AAP supports one of most important damage repair mechanisms in the body, helping to disassemble misfolded and damaged proteins. Inhibition of AAP, for example, leads to the accumulation of proteins damaged due to oxidative stress in the body, which may lead to malignant transformations. The active centre of AAP is unique: the catalytic serine (Ser587) is granted an unusual degree of conformational variability, and thus the classic serine-protease catalytic triad is loosened, alternating between active and inactive conformations. When the meropenem is in a binding monde, the active site is rearranged and AAP can no longer act as a catalyst.

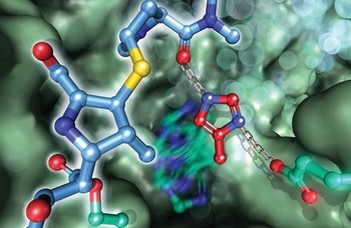
The diagram shows how the antibiotic molecule called meropenem binds to the AAP protein, as a result of which AAP can no longer act as a catalyst.(aktív=active; inaktív=inactive)
András Perczel, Head of the ELTE Department of Organic Chemistry, and his fellow researchers – Anna Kiss-Szemán, Imre Jákli, Veronika Harmat, and Dóra Karancsi Menyhárd – have verified in their current work that a commonly used penicillin-like antibiotic inhibits the functioning of the AAP protease enzyme in the human body besides fighting bacteria effectively. The study of the structure of AAP and its complex formed with the antibiotic meropenem has revealed that the antibiotic binds covalently to the active site of the enzyme and thus inhibits it irreversibly.
This is the first identified structure showing the complex of a mammalian enzyme (very similar to the human enzymes) and a penicillin derivative.
The finding above contributes to a better understanding of how AAP functions. It may also form the basis for the development of new therapeutic solutions and provide insight into the mechanisms by which antibiotics can cause side effects in human physiology.
The study was on the front page of the journal Chemical Science. It received the honorary title of “Pick of the Week” and was also included in the annual “Hot Article Collection” of the journal.
The cover page (and the front page of CS) shows the structure of the mammalian AAP protein inhibited by the antibiotic meropenem mapped by ELTE researchers.